4. Sorptiegedrag van Arseen aan illiet en gedrag in
havenslib onder aerobe en anaerobe omstandigheden.
4.1. Inleiding
Arseen komt van nature voor in het aquatische milieu door
verwering van gesteente en bodem, geothermische activiteit
(vulkanen) en lage temperatuur vervluchtiging (biologische
methylatie). Dit laatste is dus omzetting van al aanwezig As
en zorgt voor input naar de atmosfeer. Het kan ook door de
mens in het milieu gebracht worden. Dit gebeurt voornamelijk
bij de verwerking van kopererts (arsenopyriet), de verbranding
van fossiele brandstoffen (met name steenkool) en bij het
gebruik van herbiciden en pesticiden (Crecelius et al., 1975;
Brooks et al., 1982; Andreae et al., 1983; NRCC, 1978).
Hoewel arseen geen zwaar metaal is, maar een metalloïd,
wordt het meestal toch tot de zware metalen gerekend door de
overeenkomsten in giftige eigenschappen en de biogeochemische
kringloop. In tegenstelling tot andere zware metalen wordt
arseen gekarakteriseerd door een niet-kation chemie in de
waterfase. Als groep VI elementen lijken arseen en fosfor in
chemisch opzicht op elkaar. Het arsenaat-ion (As(V)) lijkt op
het voor organismen belangrijke nutriënt fosfaat en wordt
daardoor geaccumuleerd door onder andere algen, wormen,
insekten en molusken (Bryan & Gibbs, 1983; Bryan et al., 1985;
Langston, 1980, 1983; Hare et al., 1989). Arseen heeft een
negatieve invloed op de enzym werking en de energievoorziening
(A.T.P metabolisme) (NRCC, 1978), en is daarom zeer toxisch.
Het via het voedsel binnen krijgen van arseen is veel minder
schadelijk dan het inademen van vluchtige arseenspecies
(Ferguson & Gavis, 1972). De lagere organismen (algen,
bacteriën) proberen zich tegen arseen te verdedigen door
methylatie van de As-species en door reductie van As(V) naar
As(III). As(III) is weliswaar giftiger dan As(V) (Ferguson &
Gavis, 1972; Penrose, 1974) maar lijkt niet op fosfaat
waardoor het moeilijker door het organisme wordt opgenomen in
de stofwisseling (Freeman et al., 1986; Vidal & Vidal, 1980).
De methylatie van arseen-species lijkt in de natuurlijke
arseen-cyclus van sediment-water systemen van ondergeschikt
belang te zijn (McBride et al., 1978; Chau & Wong, 1978). In
oxische noch in anoxische arseen rijke sedimenten zijn
methylarseniden gevonden (Andreae, 1979; Froelich et al.,
1985). De methylatie kan door zeezout verhinderd worden (Craig
& Moreton, 1985).
Eenmaal aanwezig in het aquatische systeem kan arseen
zeer goed gerecycleerd worden (Ferguson en Gavis, 1972; Holm
et al., 1979; Agget & O'Brien, 1985). In het oxische milieu is
As(V) de voornaamste arseen-species en is voornamelijk
geassocieerd met Fe oxyhydroxiden. Onder reducerende
omstandigheden blijft As(III) mobiel in de waterkolom en in
het interstitiële water aanwezig (Agget & O'Brien, 1985;
Brannon & Patrick, 1987).
In dit onderzoek wordt het sorptiegedrag van As(III) en
As(V) aan illiet bestudeerd. Zowel kinetiek als
distributiecoëfficiënten worden bepaald. Het laatste is
gebeurd voor verschillende pH's en ionsterkten. Tenslotte is
het gedrag van As in aanwezigheid van havenslib bekeken.
4.2. Materialen en methoden
4.2.1. Materialen
Bulkoplossingen van 1000 ppm As(V) en As(III) zijn
gebruikt. As(III) is gemaakt door het oplossen van 0.3300 gram
As2O3 in 5 ml. 1 M NaOH. Hieraan wordt 130 ml. 1 M HCl
toegevoegd en vervolgens met bidest aangevuld tot 250 ml.
Deze oplossing kan minimaal
9 dagen bewaard worden
buiten de koeling, zonder
dat oxidatie naar As(V)
optreedt (zie tabel 1).
Tabel 1.
Hoeveelheid As(V) in een As(III)-oplossing.

As(V) is als 1000 ppm
standaardoplossing
voorradig. Alle andere As-
oplossingen zijn door
middel van verdunningen uit
de bulkoplossingen gemaakt.
Een 1 % oplossing van
Na-dibenzyldithiocarbamaat
is gemaakt door het
oplossen van 0,7899 gram
DBDTC in 100 ml. methanol.
Voor filtratie is gebruik gemaakt van een vacuum
filtratie systeem (Schleicher & Schöll) waarbij 0,45 æm
membraanfilters van dezelfde fabrikant worden gebruikt.
De monsters zijn gemeten op een Perkin-Elmer AAS met
holle kathode lamp.
Havenslib, afkomstig uit de Rotterdamse Waalhaven, is
bewaard in de koelcel met een laagje water op het slib.
Reducerende omstandigheden blijven op deze manier zo veel
mogelijk bewaard.
Standaard illiet is afkomstig uit Illinois.
4.2.2. Methoden
Behalve de adsorptie experimenten met havenslib, zijn
alle experimenten uitgevoerd in polyethyleen centrifugebuizen.
Er is gewerkt met 50 ml. oplossing of suspensie. Het werken in
centrifugebuizen heeft een aantal voordelen. De monsters
kunnen zeer gemakkelijk geschud en gecentrifugeerd worden.
Bovendien treedt geen adsorptie op aan de wand van de buizen
(Massee et al., 1981), waarschijnlijk omdat As als anion
(As(V)) of als neutraal molecuul (As(III)) in oplossing voor
komt. Per analyse wordt één buis gebruikt.
De adsorptie experimenten met havenslib zijn uitgevoerd
in een driehals rondbodemkolf. Op deze manier is het mogelijk
om een zuurstofloos milieu te krijgen, door N2-gas door de
oplossing te borrelen. Een snelle omschakeling naar oxiderende
omstandigheden kan worden verkregen door met perslucht te
doorborrelen. Het is zeer eenvoudig om op bepaalde tijden een
monster te nemen. Hiervoor hoeft het roeren niet gestopt te
worden, zodat de homogeniteit van de suspensie gewaarborgd is.
Een sequentiële extractie is uitgevoerd op twee
slibmonsters. Eén van de monsters is zoveel mogelijk anoxisch
gehouden. De extractie op het andere monster is onder oxische
omstandigheden uitgevoerd.
De extractie bestaat uit acht stappen. In de eerste stap
worden alle geadsorbeerde kationen vrijgemaakt met
ammoniumacetaat. De volgende stap is het vrijmaken van amorfe
ijzeroxyhydroxiden met Chester-Hughes reagens (een mengsel van
azijnzuur en hydroxylamine hydrochloride). Hierna worden alle
kristallijne ijzeroxyhydroxiden met ammoniumoxalaat uit het
monster verwijderd. Organische stof wordt vervolgens
verwijderd met waterstofperoxide, waarna weer ammoniumacetaat
aan het monster wordt toegevoegd om de eventueel geadsorbeerde
ionen weer in oplossing te brengen. Met zoutzuur worden
carbonaten en fosfaten (onder andere apatiet) in oplossing
gebracht. Silicaten worden vrijgemaakt met een mengsel van
waterstoffluoride en water. Met salpeterzuur kunnen dan de
ijzersulfiden in oplossing worden gebracht. De laatste stap is
een totaalontsluiting van het residu met waterstoffluoride en
mengzuur (perchloorzuur, salpeterzuur en water).
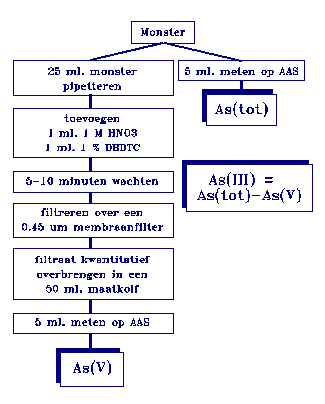
Figuur 1.
Scheidingsschema arseen.
Alle monsters zijn gecentrifugeerd (10 minuten bij 2500
toeren per minuut). Later zou blijken dat centrifugeren niet
voldoende was (zie figuur 3). De monsters zijn toen ook nog
gefiltreerd.
As wordt uit het filtraat vrijgemaakt door middel van de
boorhydride-methode. Dit houdt in dat As als gasvormig
arseenhydride een kwartsbuis wordt binnengeblazen. Deze
kwartsbuis is aangebracht in een AAS met een holle kathode
lamp. Het absorptie signaal wordt zichtbaar gemaakt op een
recorder. De piekhoogte is recht evenredig met de
concentratie. Het signaal is ook naar een computer gestuurd,
zodat piekoppervlakken bepaald kunnen worden.
Nadeel van deze methode is dat totaal As wordt bepaald.
Om As(III) en As(V) afzonderlijk te bepalen, is gebruik
gemaakt van de scheidingsmethode van Van Elteren et al.
(1989). Aan het monster wordt een overmaat DBDTC toegevoegd.
Deze stof is in water slecht oplosbaar. Complexvorming van
As(III) en vast DBDTC vindt plaats. Doordat DBDTC zo slecht
oplosbaar is in water, vlokt het direct uit en is toevoeging
van een drager niet nodig (Linder et al. 1978).
Na het neerslaan van As(III) kan het mengsel gefiltreerd
worden. Het filtraat bevat dan uitsluitend As(V). Door dit
filtraat te meten in een monster waarin zowel As(III) als
As(V) aanwezig zijn, kan de hoeveelheid As(III) berekend
worden (zie figuur 1). Aangenomen wordt dat organische As-
species niet of in verwaarloosbaar kleine hoeveelheden voor
komen.
4.3. Resultaten en discussie
4.3.1. Testen van de scheidingsmethode
De scheiding met DBDTC is in eerste instantie ontwikkeld
om As, samen met een aantal metalen, in een vaste fase te
krijgen, zodat deze via XRF gemeten kon worden (Linder et al,
1978). Van Elteren et al. (1989) optimaliseerden de coprecipitatie van As voor gebruik
met INAA. De boorhydride-methode
meet totaal As in oplossing. Daarom is gekeken of de manipulaties met het monster
(toevoeging van zuur, methanol, etc.)
geen problemen met de meting geven.
In eerste instantie is een
scheiding toegepast op een aantal As(III)-oplossingen. Bij
het scheiden moet alle As(III)
uit de oplossing worden verwijderd. Het filtraat mag dan geen
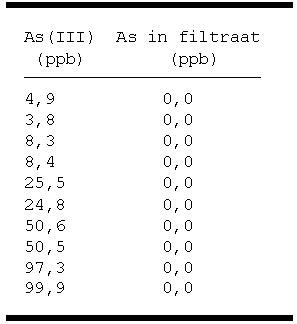
As meer bevatten. Uit tabel 2
blijkt dat dit inderdaad het
geval is. Dit betekent dat,
voor concentraties tot 100 ppb,
alle As(III) uit de oplossing
wordt afgevangen door DBDTC.
Tabel 2.
Scheiden van As(III)-oplossingen.
Een volgende stap is om
een aantal mengsels te scheiden. Negen oplossingen zijn
gemaakt door telkens 25 ml. van
een As(III)-oplossing met een
bepaalde concentratie toe te
voegen aan 25 ml. van een
As(V)-oplossing van dezelfde
concentratie. Deze mengsels zijn gescheiden. Daarna zijn de
filters opgelost in geconcentreerd zoutzuur. Per mengsel zijn
dus drie metingen uitgevoerd: een totaal monster, het filtraat
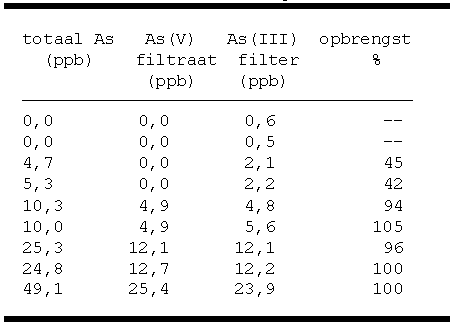
(As(V)) en het opgeloste filter (As(III)). Dit geeft de mogelijkheid om de opbrengst van
de scheiding te berekenen, met
As(III) + As(V) = As(totaal). Tabel 3 laat zien dat de opbrengst rond de 100 % ligt voor
concentraties groter dan 5
ppb. De uitschieters 94 en 105 % kunnen verklaard worden door
de fout in de boorhydride-methode. Per meting moet rekening
worden gehouden met een fout van maximaal 5 %.
Tabel 3.
Scheiden van As mengsels.
Voor concentraties lager dan 5 ppb wordt het moeilijk om
het filtraat nauwkeurig te meten. De concentratie van As(V) in
de meetoplossing wordt twee keer zo klein bij het kwantitatief
overbrengen (figuur 1). De oorspronkelijke 25 ml. monster komt
uiteindelijk in 50 ml. meetoplossing terecht. Bij afnemende
concentratie wordt de fout in de meting snel groter. Bij
concentraties lager dan 2 ppb wordt geen significant signaal
meer verkregen. Voor deze kleine concentraties kan afgeweken
worden van de standaardprocedure. Het is dan zinvol om het
filter op te lossen. Verdunning is hier niet aan de orde. Door
zo weinig mogelijk zoutzuur te gebruiken, kan zelfs tot concentraties lager dan 2 ppb
gemeten worden. De ondergrens wordt
in dit geval bepaald door de oplosbaarheid van het filter.
Er is weinig verschil tussen de resultaten verkregen met
piekhoogten en piekoppervlakken. Alleen wanneer er zeer asymmetrische pieken zijn is
het nauwkeuriger om de piekoppervlakte te bepalen. In dit onderzoek is zo veel mogelijk
gebruik
gemaakt van piekoppervlakken.
Wanneer, behalve As, ook aanzienlijke concentraties van
bepaalde metalen
in de oplossing
voorkomen (onder
andere Fe en
Mn), dan levert
scheiding problemen op. Deze
metalen coprecipiteren ook met
DBDTC (Linder et
al., 1978). Dit
blijkt uit de
kleur van het
neerslag op het
filter. Het coprecipitaat met
As heeft een
witte/gele
kleur. Bij de
diverse sequentiële extractie stappen varieerden de kleuren
van licht bruin tot rood en van donker bruin tot zwart. Bij
scheiding van de oplossing verdwijnt alle As uit de oplossing
(zie ook het paragraaf 4 van dit hoofdstuk). Het is niet
aannemelijk dat er alleen As(III) aanwezig was, zeker niet in
het oxische monster. Twee mogelijke verklaringen kunnen voor
dit verschijnsel gegeven worden. Allereerst kan er behalve een
coprecipitaat met DBDTC ook nog een ander neerslag gevormd
worden (bijvoorbeeld Fe oxyhydroxiden, waarmee As ook neerslaat). Dit is echter
onwaarschijnlijk omdat voordat het DBDTC
wordt toegevoegd de oplossing wordt aangezuurd. De tweede
verklaring is dat adsorptie van As(V) optreedt aan de gecoprecipiteerde metalen. Dit zou
plaats kunnen vinden tijdens de
wachttijd voor het filtreren.
Uit het bovenstaande wordt duidelijk dat de scheidingsmethode, in combinatie met de
boorhydride-methode, goed bruikbaar is, mits er weinig andere metalen in de oplossing
voorkomen. De scheiding is dan volledig, en de opbrengst is ongeveer
100 %. Bij lage concentraties is het beter om het filter op te
lossen, in plaats van het filtraat te meten.
4.3.2. Adsorptie van As aan illiet, kinetiek
Er is weinig tot geen literatuur te vinden die handelt
over de adsorptie van As
aan kleimineralen. Dit
is vreemd omdat kleimineralen overal ter wereld voor komen en van
grote invloed zijn op de
eigenschappen van bodem
of sediment (onder andere adsorptie-kenmerken). Hier is gekozen
voor het kleimineraal
illiet (ideale formule:
K0.7Al2(Si,Al)4O10(OH)2)
omdat illiet op gematigde breedten één van de
belangrijkste kleimineralen is (zie figuur 2).
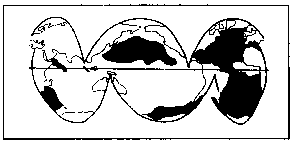
Figuur 2.
Verdeling van illiet in de oceanen (naar Griffin et al., 1968).
Immers de verdeling van kleimineralen wordt onder andere
bepaald door het klimaat (Gillot, 1968). Hoewel figuur 2 de
distributie in de oceanen geeft, komt die overeen met de
verdeling op de continenten (Pannekoek, 1982).
Allereerst is gekeken naar de kinetiek van de adsorptiereacties. Na enkele testen is
gekozen voor een suspensiegehalte van 0.5 g/l illiet, en een initiële arseen
concentra-
tie van ongeveer 50 ppb. Het suspensie-gehalte is hoger dan de
gehalten in rivieren. Voor de Rijn, bijvoorbeeld, geven Van
der Weijden & Middelburg (1989) gehalten van gemiddeld 57 ñ 27
mg/l. Uit de inleidende proeven kwam een onduidelijk beeld
naar voren (zie figuur 3). De pH van de suspensie is ongeveer
6,5, buffering is niet toegepast. Duplo's liggen meestal dicht
bij elkaar, maar een
continue afname van de
As(V)-concentratie is
nauwelijks zichtbaar.
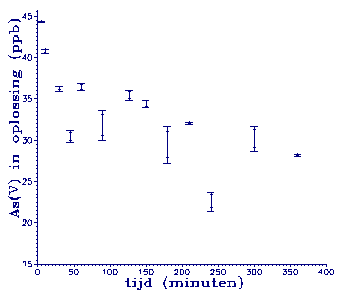
Figuur 3.
Adsorptie van As(V) aan illiet. De monsters zijn alleen gecentrifugeerd.
Vermoed werd dat dit
veroorzaakt werd door de
aanwezigheid van illietdeeltjes met daaraan
geadsorbeerd As in de
oplossing na centrifugeren. Tijdens de analyse
voorbereiding komt dit
As geheel of gedeeltelijk weer in oplossing
en verstoort de meting.
Besloten is toen om de
monsters na centrifugeren ook nog te filtreren. Het vermoeden bleek
juist te zijn, want op
het filter bleef illiet
achter. Adsorptie van As
aan het filter treedt
niet op (Massee et al., 1981).
De resultaten van de adsorptie van As(III) en As(V) zijn
gegeven in figuur 4.
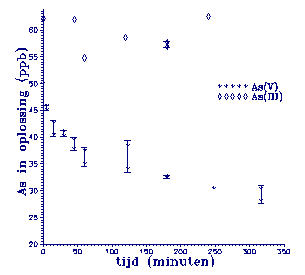
Figuur 4.
Adsorptie van As(V) en As(III) aan illiet.
De initiële concentraties zijn 50 ppb en
62 ppb voor As(V) respectievelijk As(III). Wat op valt is het
grillige gedrag van As(III). Een significante afname is niet
waarneembaar. Op t=248 minuten is er niets geadsorbeerd. Op
eerdere tijdstippen lijkt tot 7.5 ppb uit de oplossing verdwenen te zijn. Deze afname
verloopt niet volgens een continu
aflopende curve, zoals bij As(V) wel het geval is. Dit slechte
adsorptiegedrag is te wijten aan het feit dat As(III) bij de
experimentele pH (þ 6,5) als neutraal HAsO2 in de oplossing
zit. Adsorptie is dan moeilijk en treedt nauwelijks op. Oxidatie van As(III) wordt niet
bevorderd door illiet (Oscarson
et al., 1981a).
As(V) daarentegen, laat een duidelijke concentratie
afname in de oplossing zien. De adsorptiereactie kan als volgt
worden weergegeven:
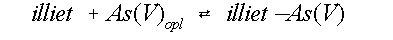 (1)
(1)
met illiet (in de reactievergelijking 1) is de hoeveelheid
adsorptieoppervlak, As(V)opl is de hoeveelheid As(V) in oplossing en illietÄAs(V)
is de
hoeveelheid geadsorbeerd As. De
reactie bestaat dus uit twee deelreacties die tegelijkertijd
verlopen: een adsorptie- en een desorptie-reactie.
Nu kan de snelheidskonstante berekend worden. Hierbij
wordt gebruik gemaakt van de isolatie-methode (Lasaga & Kirkpatrick, 1981). Alleen de
concentratie afname van As(V) wordt
bekeken, de hoeveelheid adsorptie-oppervlak wordt konstant
verondersteld. Een eerste orde reactie blijft dan over. De
vergelijking daarvoor is:
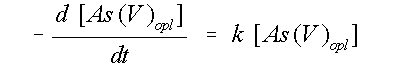 (2)
(2)
Na integratie gaat vergelijking 2 over in:
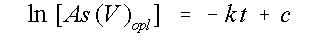 (3)
(3)
waarin [As(V)opl] de
concentratie As(V)
in oplossing is op
tijdstip t, k de
snelheidskonstante
en c een konstante
(in het ideale
geval is c gelijk
aan de initiële
As(V)-concentratie). Als de aanname van een eerste
orde reactie correct is, dan moet
de grafiek van ln
[As(V)opl] tegen t
een rechte lijn op
leveren met helling
-k. In dit geval is
de snelheidskonstante een netto k,
immers de snelheid
van de reactie
wordt bepaald door zowel adsorptie als desorptie. De data zijn
geplot in figuur 5.
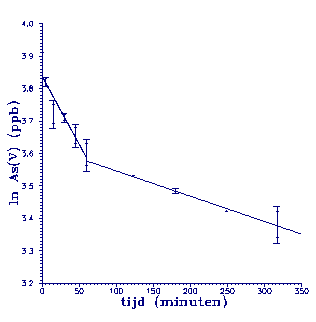
Figuur 5.
Eerste orde benadering. In de grafiek zijn twee lijnen getrokken die de twee
snelheidskonstanten aangeven.
In plaats van één rechte lijn, kunnen in
de grafiek twee rechte lijnen getrokken worden. Eén voor het
traject 0 tot 60 minuten en één voor het traject 60 tot 350
minuten. Dit betekent dat de snelheidskonstante slechts binnen
een bepaald tijdsinterval geldig is. Het verschijnsel van twee
snelheidskonstanten is niet ongewoon in een heterogeen systeem. Oscarson et al. (1983b)
vinden dit voor de oxidatie van
As(III) door mangaanoxiden, Jurinak & Griffin (1972) voor de
adsorptie van nitraat aan calciumcarbonaat.
De konstanten voor beide trajecten zijn 4,19 * 10-3 min-1
(0-60 minuten) en 7,79 * 10-4 min-1 (60-350 minuten). Dit houdt
in dat de eerste konstante 5,4 keer zo groot is als de tweede.
Dit verschil kan verklaard worden door het belangrijker worden
van desorptie processen. Deze processen zijn vaak langzamer
dan adsorptie processen (Jurinak & Griffin, 1972) en gaan pas
een rol spelen als de concentratie van het adsorberende species (in dit geval As) laag
genoeg is. Voor As(V) blijkt dat
desorptie in het eerste uur geen rol speelt. Daarna neemt de
adsorptie snelheid af, dat wil zeggen, de netto adsorptie
snelheid. Het is goed mogelijk dat de adsorptie snelheid konstant blijft of zelfs toeneemt
(wat niet waarschijnlijk is).
Omdat de desorptie snelheid toeneemt, wordt de netto snelheid
lager.
Uit het bovenstaande kan worden geconcludeerd dat adsorptie van As aan
kleimineralen een moeizame zaak is.
As(III), de meest giftige vorm, adsorbeert helemaal niet.
Adsorptie van As(V) is redelijk snel gedurende het eerste uur,
30 % van de toegevoegde hoeveelheid, maar neemt daarna sterk
af, minder dan 20 % over een vijf keer zo lang tijdsinterval.
4.3.3. Adsorptie van As(V) aan illiet,
distributie-coëfficiënten
Voor verschillende pH's is de distributie-coëfficiënt
bepaald. De monsters, met een suspensie gehalte van 0,5
gram/liter illiet, hadden een equilibratie tijd van tien
dagen. Extrapolatie van de adsorptie-curve in figuur 4 toont
aan dat na tien dagen zeker geen adsorptie meer optreedt. De
pH is constant gehouden met een NaHCO3 buffer. De ionsterkte is
vastgelegd op 0,003 M met CaCl2. Tevens is een experiment in
Middellands Zeewater uitgevoerd (pH = 8,0, I = 0,7).
De distributie-coëfficiënt geeft de verhouding geadsorbeerd
As : As in oplossing.
Voor de berekening is van de
volgende formule gebruik gemaakt:
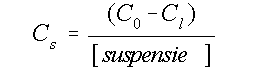 (4)
(4)
Hierin is Cs de hoeveelheid geadsorbeerd As(V) per gram illiet,
C0 de toegevoegde concentratie As(V) aan de oplossing,
Cl de concentratie van As(V)
na equilibratie en [suspensie] is het aantal grammen
illiet per liter oplossing.
De distributie-coëfficiënt Kd wordt dan:
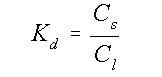 (5)
(5)
Figuur 6 toont de verschillende adsorptie isothermen.
Meteen wordt duidelijk dat er
verschillen zijn tussen de
diverse pH's. De distributie-coëfficiënten zijn berekend en
gegeven in tabel 4.
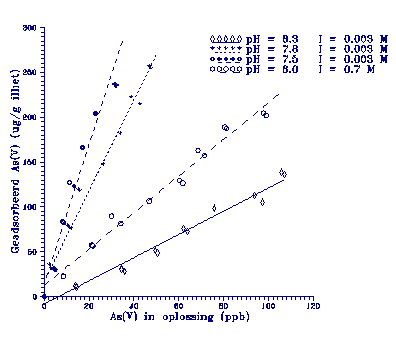
Figuur 6.
Adsorptie isothermen voor verschillende pH's en ionsterkten.
De equilibratietijd is 10 dagen.
Bij lage ionsterkte is er een toename van
de Kd te zien bij afnemende pH. Dit betekent dat bij afnemende
pH meer As(V) adsorbeert. De reden hiervoor moet gezocht
worden in de pH-afhankelijke speciatie van As(V) (zie figuur
7). Het pH-traject 7,5 tot 8,3 laat een afname zien van de
H2AsO4- bijdrage ten gunste van HAsO42- (de pK van de reactie
waar H2SO4- een proton afsplitst is 6,98). De Kd neemt dus af
bij een toenemend HAsO42- aandeel en een afnemend H2AsO4- aandeel
(zie ook tabel 4). Geconcludeerd kan worden dat dus voornamelijk H2AsO4- adsorbeert.
Tabel 4.
Distributiecoefficienten bij vershilllende pH's en ionsterkten. Bij elke pH is ook de
hoeveelheid H2AS)4- gegeven.

Voor fosfaat, vergelijkbaar met arsenaat
wat betreft de opbouw,
is een zelfde beeld
geconstateerd. Een
maximum adsorptie vindt
plaats in de pH-range
van 4 tot 7 (Beek & Van
Riemsdijk, 1979). In
dit traject is H2PO4 het dominante species.
Waarom het eenwaardige
negatieve species het
best adsorbeert is eenvoudig te verklaren.
Anionen worden afgestoten door de negatieve
lading van de platen
van de kleimineralen.
Hoe negatiever het
anion hoe groter de afstoting. Een eenwaardig
negatief ion kan dan
nog het gemakkelijkst in de buurt van het kleimineraal komen
om daar te adsorberen aan de positief geladen randen.
Ook de ionsterkte is van invloed op de distributie-coëfficiënt. Voor
zeewater met een
pH van 8,0 is de Kd 2,17. Indien
de ionsterkte geen invloed heeft op de Kd, dan zou deze ongeveer 3,6 moeten zijn
(berekend met data uit tabel 4). De Kd is
echter duidelijk lager, er adsorbeert dus minder As(V). Dit
wordt veroorzaakt door de grote competitie van arsenaat met
andere anionen in het zeewater.
Het is mogelijk dat bij een toename van de ionsterkte de
adsorptie van As(V) beter wordt. De kationen adsorberen aan
het negatieve oppervlak en 'neutraliseren' op die manier de
negatieve lading. Hierdoor treedt minder afstoting op, zodat
adsorptie gemakkelijker wordt. Bowden (1973) en De Haan (1965)
hebben aangetoond dat, voor fosfaat adsorptie aan montmorilloniet, de adsorptie groter
wordt bij toenemende ionsterkte.
In zeewater zijn de concentraties aan anionen echter dermate
groot dat het positieve effect van de kationen te niet wordt
gedaan.
4.3.4. Havenslib
Tenslotte zijn er enige experimenten met havenslib uitgevoerd. Eerst is een
sequentiële extractie uitgevoerd op een
oxisch en een anoxisch monster. Hiermee kan bepaald worden in
welke minerale fasen As in het slib voorkomt. Daarna is gekeken of het anoxische slib,
onder anaerobe omstandigheden, As
afstaat aan demi-water. Dit laatste is bereikt door demi-water
anderhalf uur te doorborrelen met stikstof-gas. Na een uur
roeren is een monster genomen. Al het eventueel aanwezige As
in het poriënwater en zwak geadsorbeerd As is dan verspreid
door de oplossing. Vervolgens is met perslucht doorgeborreld
om te kijken hoe de concentratie in de oplossing verandert
onder aerobe omstandigheden. Het derde experiment is kwa uitvoering vrijwel gelijk,
echter nu wordt As toegevoegd aan de
suspensie, zodat de adsorptie aan geoxideerd havenslib bekeken
kan worden.
Voordat de resultaten besproken worden, is het zinvol om
de mogelijke processen afzonderlijk te bespreken. Het is
duidelijk dat in een anoxisch sediment of slib een aantal
processen (oxidatie/reductie, adsorptie) een rol spelen. Deze
processen overlappen elkaar gedeeltelijk en veranderen als het
slib in een oxisch milieu terecht komt.
Arseen kent twee verschillende oxidatie toestanden,
waarvan As(III) aanzienlijk giftiger is dan As(V). Belangrijk
is dus in welke vorm As in oplossing voor komt. De verhouding
As(III)/As(V) wordt bepaald door de Eh. Deze afhankelijkheid
wordt uitgedrukt in de volgende vergelijking (Kersten, 1980):
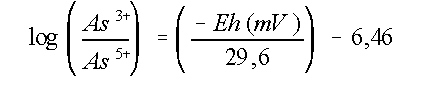 (6)
(6)
Deze vergelijking geldt voor een pH van 7,3, typisch voor
anoxische sedimenten. Verschillende onderzoeken melden een
As(III)/As(V) ratio variërend van 0,1 tot 10 (Johnson, 1972).
Dit komt overeen met Eh-waarden tussen -220 en -160 mV. Theoretisch kan een
Eh-verschil van 60 mV een totale verandering
van het dominante species tot stand brengen.
Een belangrijk proces is dan ook de oxidatie van As(III)
naar As(V). Zuurstofrijk water alleen is echter onvoldoende om
de oxidatie reactie te laten verlopen (zie tabel 1; Oscarson
et al, 1980, 1981b). Een thermodynamisch gunstige reactie is
oxidatie met behulp van mangaan(IV)oxiden (þG0 = -86,9 kJ/mol).
Oscarson et al. (1983a) geven dit proces weer met de volgende
reactievergelijkingen:
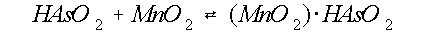 (7)
(7)
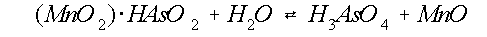 (8)
(8)
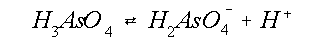 (9)
(9)
 (10)
(10)
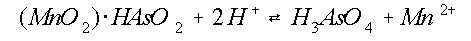 (11)
(11)
De eerste stap is de adsorptie van arseniet aan Mn(IV)-
oxide (vergelijking 7). Vervolgens wordt HAsO2 geoxideerd naar
H3AsO4 en Mn(IV) wordt Mn(II), waarbij ook zuurstof overdracht
plaats vindt (vergelijking 8). Arsenaat dissocieert, afhankelijk van de pH, in verschillende
hoeveelheden H2AsO4- en HAsO42-,
waarbij H+ in oplossing komt (vergelijking 9 en 10, figuur 7).
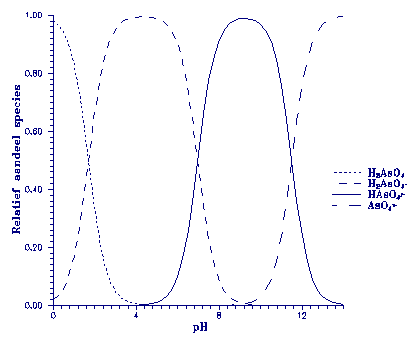
Figuur 7.
Speciatie van As(V) als functie van pH.
De vrijgekomen protonen kunnen direct reageren met geadsorbeerd arseniet om H3AsO4
en Mn2+ te vormen (vergelijking 11).
Gedeeltelijk komt het gereduceerde Mn dus in oplossing. Eventueel kan het daar weer
geoxideerd worden, zodat het weer in
het oxidatie proces kan meedoen.
Oscarson et al. (1983b) hebben verschillende mangaan(IV)-
oxiden onderzocht. Hieruit komt naar voren dat het algemeen
voorkomende birnessiet (þ-MnO2) het meest effectief is voor de
oxidatie. Dit mineraal komt voor als discrete korrels of als
coating op andere korrels. Indien de Mn-korrels zelf een
coating hebben, van bijvoorbeeld Al-oxide, dan kunnen de
vrijkomende protonen deze coating dusdanig aantasten dat het
Mn-oxide aan de oxidatie deel kan nemen.
Arseniet kan ook geoxideerd worden door ijzer(III)oxiden.
Ook deze reactie is thermodynamisch gunstig (þG0 = -32,6
kJ/mol). Oscarson et al. (1981b) vinden geen oxidatie van
As(III) in de aanwezigheid van Fe2O3. Ze concluderen hieruit
dat de kinetiek van de reactie te langzaam is om een rol van
betekenis te spelen. Belzile et al. (1989) hebben het tegendeel aangetoond voor
ijzeroxyhydroxiden. Binnen een paar dagen
wordt minimaal 50 % van het aanwezige arseniet geoxideerd. Dit
zou betekenen dat de oxidatie capaciteit sterk afhankelijk is
van de vorm waarin ijzer(III) voor komt. In oxide-vorm treedt
geen oxidatie van As op, in de oxyhydroxide-vorm wel.
Voor een aantal andere mineralen is onderzocht of ze
belangrijk zijn bij de oxidatie van arseniet (Oscarson et al,
1981a). Montmorilloniet, kaoliniet, illiet, vermiculiet,
smectiet, mikroklien, orthoklaas en calciet zijn toegevoegd
aan een As(III)-oplossing. Na 48 uur is geen As(V) in de
oplossing gemeten. Deze silikaten en carbonaten spelen dus
geen rol bij de oxidatie van arseniet.
Oxidatie kan ook optreden in de waterkolom. Bacteriën
zijn hier voor verantwoordelijk (Johnson & Pilson, 1975).
Johnson & Pilson (1975) hebben verschillende parameters gevarieerd tijdens
het oxidatie
proces. Een temperatuur verhoging van 10øC betekent een verdubbeling
van de snelheid. De
zuurstof concentratie heeft nauwelijks invloed, de saliniteit
echter wel. Bij toenemende saliniteit wordt de snelheid groter.
Ook de aanwezigheid van
zonlicht versnelt de oxidatie
(fotochemische oxidatie). De natuurlijke oxidatie snelheid
(zowel organisch als anorganisch) is ongeveer 0,023 æmol
As(III) l-1 jaar-1 in zeewater.
De omgekeerde reactie, reductie van arsenaat, kan natuurlijk ook plaats vinden. In de
waterkolom vindt reductie onder
invloed van bacteriën plaats (Johnson, 1972; Freeman et al.,
1986). Eerstgenoemde heeft met behulp van laboratorium experimenten een arsenaat
reductie snelheid bepaald van ongeveer
10-11 æmol cel-1 minuut-1, een langzame reactie dus.
In anoxisch sediment verloopt het reductie proces beter.
Ook hier zijn bacteriën van belang (Freeman et al., 1986),
maar een voldoende lage Eh is belangrijker. De reductie van
arsenaat is nooit volledig, in poriënwater wordt altijd nog
As(V) gevonden. Dit wordt veroorzaakt doordat de Eh niet
altijd optimaal is voor reductie. De kinetiek is te langzaam
en omwoeling van het sediment zorgt weer voor gedeeltelijke
oxidatie. Het laatste proces kan plaats vinden tijdens stormen
of wordt veroorzaakt door stromingen (Huang et al., 1988).
Reductie naar arseniet gaat samen met mobilisatie van As
in het sediment. Ook Fe(III) en Mn(IV) worden gereduceerd.
Volgens Agget & Kriegman (1988) worden eerst Fe en Mn gereduceerd, waardoor het
geadsorbeerde en gecoprecipiteerde arsenaat in oplossing komt en op zijn beurt
gereduceerd wordt.
Holm (1988) beweert dat de volgorde omgekeerd is: eerst reductie van As(V) en daarna
pas van Fe en Mn. Hij heeft een reductie snelheid berekend door te kijken naar de
hoeveelheid
vrijkomend As(III) uit een sediment. Aangenomen wordt dat dit
per tijdseenheid even veel is als er arsenaat gereduceerd
wordt. De snelheid is 10-5 g kg-1 dag-1 ofwel 2,1 mg m-2 dag-1.
Dit is in overeenstemming met waarden die gevonden zijn door
diverse andere onderzoekers.
Een belangrijk proces is adsorptie van As. Eerder is al
opgemerkt dat er nauwelijks literatuur te vinden is over
adsorptie aan kleimineralen. Adsorptie aan organische stof
speelt een ondergeschikte rol (Salomons & Förstner, 1980).
Daarom wordt hier alleen de adsorptie aan metaaloxiden besproken.
Mangaan(IV)oxiden oxideren niet alleen arseniet, maar
kunnen ook As adsorberen. Oscarson et al. (1983b) hebben de
adsorptie aan verschillende mangaanoxiden onderzocht. Het
blijkt dat As(III) in het algemeen beter adsorbeert dan As(V).
Cryptomelaan (þ-MnO2) heeft de grootste adsorptiecapaciteit. De
geringe hoeveelheid As(V) die adsorbeert is een gevolg van het
lage Point of Zero Charge (PZC). Dit ligt bij een pH van
ongeveer 2. Bij neutrale pH waarden is het oxide oppervlak
negatief geladen, zodat voor adsorptie van een arsenaat ion
een hoge energiedrempel moet worden overwonnen. Adsorptie van
As(V) is wel gemakkelijk als arseniet aan het oxide oppervlak
wordt geoxideerd. De negatieve oppervlakte lading speelt dan
geen rol omdat As(V) bij vorming meteen geadsorbeerd wordt.
Dit wordt bevestigd door Oscarson et al. (1981b).
Het percentage As dat geadsorbeerd is aan carbonaten,
organische stof en sesquioxiden, bedraagt 68 tot 84 % in
pelagische sedimenten en 66 tot 70 % in estuariene sedimenten.
42 tot 58 % zit aan Fe-sesquioxiden (Maher, 1984). IJzeroxiden
hebben een positieve oppervlaktelading tot pH þ 8,5. Dit
betekent dat adsorptie over een breed pH-trajekt kan plaats
vinden. Oscarson et al. (1981b) laten zien dat arseniet beter
adsorbeert aan ijzeroxiden dan arsenaat. Binnen 72 uur neemt
de concentratie af met bijna 100 ppm As(III) in aanwezigheid
van 100 mg Fe2O3. Voor As(V) ligt dit op bijna 50 ppm.
De adsorptie aan Fe-oxyhydroxiden is bestudeerd door
Belzile et al. (1989). Dit proces kan met de volgende formules
worden beschreven:
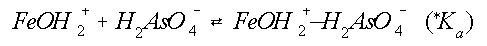 (12)
(12)
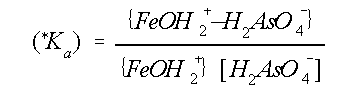 (13)
(13)
(*Ka) is de ogenschijnlijke evenwichtskonstante. {FeOH2+} en
{FeOH2+ÄH2AsO4-} staan voor de concentratie van vrije oppervlakte sites van
het
Fe-oxyhydroxide respectievelijk sites die
bezet zijn door As. Aangenomen wordt dat {FeOH2+ÄH2AsO4-} þ
{FeOH2+}. Verder geldt:
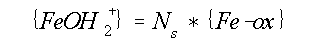 (14)
(14)
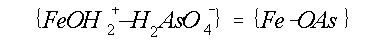 (15)
(15)
waarin Ns de dichtheid van sites is. {Fe-ox} en {Fe-OAs} staan
voor concentraties van diagenetisch ijzer oxyhydroxide respectievelijk geassocieerd As.
Combinatie van de vergelijkingen
13, 14 en 15 levert:
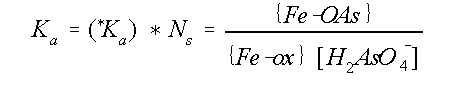 (16)
(16)
Figuur 8 toont curven die berekend zijn uit adsorptie data met
authigene Fe-oxyhydroxiden.
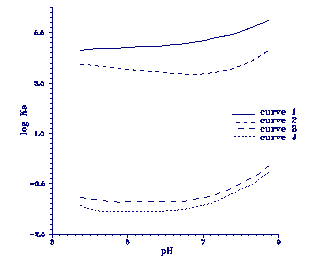
Figuur 8.
Evenwichtskonstanten Ka, voor adsorptie van As aan Fe-oxyhydroxiden als functie van de
pH (naar Belzile et al., 198-9.
Curve 1 en 2 zijn bepaald met
amorf ijzer oxyhydroxide met As concentraties van 0,65 respectievelijk 41,7 æM.
Curve 3
en 4 zijn bepaald met goethiet met
As concentraties van 1,07 respectievelijk 0,53 æM. Hieruit
blijkt dat goethiet een ondergeschikte rol speelt in het adsorptie proces. De *Ka is vrijwel
pH-onafhankelijk, zoals ook
al bleek uit vergelijking 16. Een verandering van de H2AsO4-concentratie, veroorzaakt
door een pH-verandering van het systeem, wordt opgevangen door een verandering in de
hoeveelheid
geadsorbeerd As (vergelijking 15). Velddata komen overeen met de adsorptie aan amorf
Fe-oxyhydroxide.
Het onderzoek van Belzile et al. (1989) toont ook aan dat As(III) geoxideerd wordt door
Fe oxyhydroxiden. Alleen As(V) adsorbeert, dit in tegenstelling tot de resultaten van
Oscarson et al. (1981b).
Als het sediment in een anaeroob milieu terecht komt, wordt As(V)
gereduceerd en komt dan in oplossing. Vervolgens kan zowel
opwaartse als neerwaartse diffusie optreden. Bij opwaartse
diffusie kan arseniet weer geoxideerd worden, waarna wederom
adsorptie kan plaats vinden. As(III) kan ook in de waterkolom
terecht komen als vrij ion (Belzile & Tessier, 1990). Bij
neerwaartse diffusie kan As2S3 worden gevormd of arseniet kan
neerslaan met ijzersulfiden (Belzile & Lebel, 1986). Tijdens
pyrietvorming in suboxische sedimenten, kan Fe vervangen worden
door As. De verhouding Fe/As is ongeveer 1000.
As kan ook coprecipiteren met Fe-oxiden. Maher (1984) beweert dat
een aanzienlijk deel van het As in het sediment zich bevindt in
goed gekristalliseerde ijzeroxiden.
De resultaten van de sequentiële extractie zijn gegeven in tabel
5.
Tabel 5.
Sequentiële extractie.
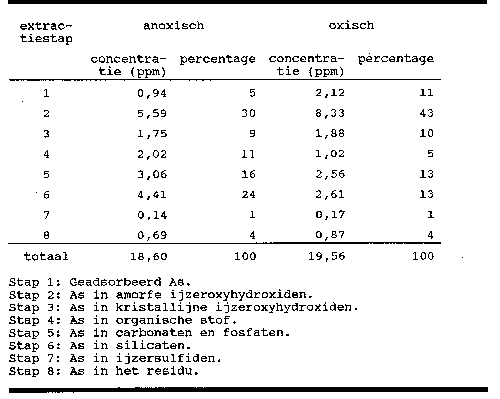
Zoals verwacht kon worden zit een zeer aanzienlijk deel van
het arseen in de ijzeroxyhydroxiden (39 en 53 % voor het
anoxische respectievelijk oxische monster). Opvallend is het hoge
aandeel van de amorfe ijzeroxyhydroxiden (stap 2). De hoeveelheid
As in kristallijne ijzeroxyhydroxiden (stap 3) is ongeveer gelijk
aan de hoeveelheid geadsorbeerd As (stap 1) en de hoeveelheid As
in organische stof (stap 4). Alle fasen bevatten ongeveer 10 %
van het totaal. Iets hogere waarden worden gevonden in de fosfaat
en carbonaat fase (stap 5) alsmede in de silicaat fase (stap 6).
Het anoxische monster heeft een uitschieter in de silicaat fase
met 24 %. In ijzersulfiden (stap 7) blijkt weinig As aanwezig te
zijn. Voor het oxische monster is dit logisch, omdat er geen
ijzersulfiden aanwezig zijn. Voor het anoxische monster werd een
hoger gehalte verwacht. Een verklaring voor deze lage hoeveelheid
kan gevonden worden in het feit dat het bij stap 4 gebruikte
waterstofperoxyde de ijzersulfiden oxideert. Deze extractie
volgorde zou dan niet bruikbaar zijn.
Het tweede experiment met havenslib geeft aan dat weinig As vrij
komt onder anaerobe omstandigheden (zie tabel 6).
Tabel 6.
As concentratie in oplossing, in aanwezigheid van havenslib.
Op t=0 minuten wordt overgegaan van anaerobe naar aerobe omstandigheden.

Uit de
sequentiële extractie blijkt dat gemiddeld 19 ppm As in het slib
aanwezig is. Hier is gewerkt met een suspensiegehalte van 1,53
gram/l slib. Dit komt overeen met 29,1 ppb As in suspensie. Er
wordt slechts 1,5 ppb gemeten in oplossing (tabel 6). Een klein
gedeelte ( 5 %) van het As komt dus maar in oplossing. Dit is
precies even veel als de geadsorbeerde hoeveelheid As van het
anoxische monster (zie tabel 5). De adsorbeerde fase is dus de
enige fase die gemakkelijk in oplossing komt.
Op t = 0 minuten is de suspensie nog anoxisch. Het eerste monster
geeft de concentraties in het anoxisch milieu. De kleine
hoeveelheid As wordt zeer snel verwijderd (binnen 15 minuten)
wanneer een oxisch milieu wordt gecreëerd. Deze verwijdering
kan plaats vinden door coprecipitatie net ijzeroxyhydroxiden,
adsorptie aan metaaloxiden en kleimineralen of door een
combinatie van deze processes. Dat er ijzer(hydr)oxiden neerslaan
in het havenslib, is te zien aan de kleurverandering van zwart
(anoxisch) naar bruin-geel (oxisch). Coprecipitatie met
ijzeroxyhydroxiden lijkt bevestigd te worden door de
sequentiële extractie: het oxische monster bevat meer As in
de amorfe ijzeroxyhydroxide fase. Dit moet dus het zwak gebonden
As zijn geweest wat onder anoxische omstandigheden in oplossing
komt. Het derde experiment bestaat uit drie delen (zie figuur 9).
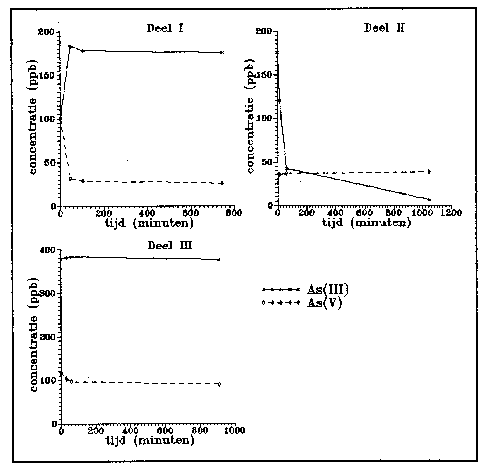
Figuur 9.
Concentraties van As(III) en As(V) als functie van de tijd.
Deel I is anoxisch, deel II en III zijn oxisch.
In het begin, deel I, is de suspensie (1,53 g/1) anoxisch.
De initiële Asconcentraties zijn 100 ppb voor zowel AS(III)
als As(V). Onder invloed van de lage Eh neemt de hoeveelheid
As(V) sterk af in de eerste 45 minuten ( 75 % verdwijnt). Al het
verdwenen AS(V) is omgezet naar As(III), adsorptie lijkt niet op
te treden. Het reductieproces verloopt steeds langzamer in de
tijd. Wanneer snel geschakeld wordt naar een oxisch milieu (deel
II), dan vindt en snelle verwijdering plaats van arseniet.
Dezelfde processen als bij het niet te achterhalen hoeveel
arseniet geoxideerd wordt, en door welk metaaloxide. Ook de
verhouding gecoprecipiteerd As/geadsorbeerd As is niet te
bepalen. As(III) wordt wel zeer effectief verwijderd (meer dan 95
%). Opvallend is het vrijwel constant blijven van de As(V)
concentratie. Na de beluchting van de suspensie loopt dit gehalte
ongeveer 10 ppb op en blijft dan op ongeveer 37 ppb steken. Een
verklaring voor dit verschijnsel kan niet gegeven worden.
Daarvoor zou meer in detail gekeken moeten worden in welke vorm
en waar het uit de oplossing verwijderde As(III) is gebleven.
Wanneer in deel III nogmaals As wordt toegevoegd (370 ppb
AsS(III) en 80 ppb As(V)), gebeurt er niet veel meer. Er is
slechts een lichte afname van As(V). Blijkbaar is de oxidatie
capaciteit van het slib niet meer aanwezig. Hier zijn twee
redenen voor aan te voeren. Allereerst kunnen de mangaanoxide
korrels, bij overschakeling van anoxische naar oxische omstan-
digheden, een coating van amorf ijzeroxyhydroxide hebben
gekregen. Vervolgens zou het ijzeroxyhydroxide nog het As(III)
kunnen oxideren. Dat dit niet gebeurt wijst op een volledige
bezetting van het oppervlak met As(V).
Adsorptie van As(V) vindt nog wel plaats (de concentratie neemt
met ongeveer 25 ppb af in oplossing), maar de bezetting van de
adsorptie plaatsen lijkt vrijwel volledig. De totale hoeveelheid
As die bij beluchting en latere adsorptie verwijderd is uit de
oplossing is 180 &#u;g/l. Dit gebeurt door 1,53 g/l havenslib,
dit betekent dat 118 mg As/g slib verwijderd is. Wanneer nu
nogmaals As toegevoegd zou worden, dan is de verwachting dat nog
maar weinig adsorptie op zal treden. De bezetting van de
adsorptie plaatsen lijkt vrijwel volledig (nog maar zeer langzame
adsorptie, terwijl toch nog 90 ppb As(V) in de oplossing aanwezig
is).
De grootste hoeveelheid As verdwijnt bij de overgang van
anoxische naar oxische omstandigheden. Waarschijnlijk wordt het
meest As gecoprecipiteerd met Fe oxyhydroxiden.
Uit deze proeven blijkt dat havenslib, voor wat As betreft,
gewoon gestort worden. Immers de kleine hoeveelheid As die vrij
kan komen wordt zeer snel weer in het slib opgenomen. Alleen als
een aanzienlijk deel van het As geadsorbeerd is (wat
onwaarschijnlijk is), is storting in zeewater niet aan te bevelen
omdat As dan desorbeert en in de waterkolom terecht komt.
Wanneer de gestorte hoeveelheden slib zeer groot zijn, kunnen
hierdoor lokaal verhoogde concentraties van As optreden. Dit is
echter een nauwelijks reële situatie.
4.4. Conclusies
De scheidingsmethode voor As(III) en As(V) met DBDTC, in
combinatie met meting volgens de boorhydride-methode, is zeer
goed bruikbaar. Voorwaarde is wel dat er geen hoge concentraties van andere metalen in
de oplossing mogen zitten. De
scheiding is volledig, in ieder geval tot 100 ppb. De standaardprocedure gaat uit van twee
metingen, een totaal monster
en het filtraat. Indien de concentratie As in het filtraat te
laag is, kan het filter opgelost worden. Bepaling van lage
concentraties is dan mogelijk (< 2 ppb).
Arseniet, een neutraal molecuul tot pH þ 9, adsorbeert
niet aan illiet. Arsenaat adsorbeert volgens een eerste orde
reactie. Er zijn twee snelheidskonstanten bepaald voor de
adsorptie reactie: 4,19 * 10-3 min-1 voor het traject van 0 tot
60 minuten, en 7,79 * 10-4 min -1 voor het traject van 60 tot
350 minuten. Na een redelijk snelle adsorptie in het eerste
uur neemt de adsorptiesnelheid daarna sterk af ten gevolge van
desorptie.
Bij adsorptie van As(V) is vooral de pH van belang. In
het pH-traject van 7,5 tot 8,3 neemt adsorptie af met toenemende pH. Tevens neemt de
H2AsO4- concentratie af. Het belangrijkste adsorberend species is dus H2AsO4-, dit in
analogie met
fosfaat waar H2PO4- het best adsorbeert. Voor een eenwaardig
negatief ion is de afstoting door de negatief geladen platen
van illiet het kleinst, dus treedt adsorptie het gemakkelijkst
op.
Ook de ionsterkte speelt een rol. Bij lagere ionsterkten
heeft een kleine verhoging een sterkere adsorptie tot gevolg.
Dit wordt veroorzaakt door 'neutralisatie' van de platen van
de kleimineralen door adsorptie van kationen. Indien de ionsterkte te groot wordt, zoals in
zeewater, neemt de adsorptie
af ten gevolge van de grotere competitie van As met andere
anionen.
In een suspensie met havenslib kunnen een aantal processen een rol spelen. De
belangrijkste zijn oxidatie van arseniet door ijzeroxyhydroxiden en mangaanoxiden, en
adsorptie
van voornamelijk arsenaat aan ijzeroxyhydroxiden. Uit de
experimenten blijkt dat As onder anaerobe condities niet
gemakkelijk vrij komt. Slechts 5 % van het in het slib aanwezige As komt in oplossing.
Dit is geadsorbeerd As zoals uit
de sequentiële extractie blijkt. Bij oxidatie van het systeem
wordt de kleine hoeveelheid As binnen 15 minuten weer verwijderd. Het meeste As wordt
verwijderd wanneer de overgang
anoxisch/oxisch op treedt. Hieruit blijkt dat coprecipitatie
met, waarschijnlijk, Fe oxyhydroxiden het belangrijkste verwijderingsmechanisme is. Dit
wordt bevestigd door de sequentiële extractie, die aantoont dat de amorfe
ijzeroxyhydroxiden
het meeste As bevatten. Kristallijne ijzeroxyhydroxiden bevatten aazienlijk lagere
hoeveelheden. Samen zijn deze twee fasen
goed voor ongeveer de helft van het As in het havenslib.
Uit deze proeven blijkt dat storting van dit havenslib
zonder problemen kan plaats vinden, voor wat betreft As. De
kleine hoeveelheid As die onder anaerobe condities vrij kan
komen, wordt vrijwel direct verwijderd wanneer het slib in een
oxisch milieu terecht komt.
4.5. Referenties
Agget, J. & Kriegman, M.R. (1988): The extent of formation of
arsenic(III) in sediment interstitial waters and its release
to hypolimnetic watrs in Lake Ohakuri. Water Research, 22,
407-411.
Aggett, J. & O'Brien, G.A. (1985): Detailed model for the
mobility of arsenic in lacustrine sediments based on measurements in Lake Ohakuri.
Environmental Science and Technology, 19, 231-238.
Andreae, M.O. (1979): Arsenic speciation in sea water and
interstitial waters: the influence of biological-chemical
interactions on the chemistry of a trace element. Limnol.
Oceanogr., 24, 440-452.
Andreae, M.O., Byrd, J.T. & Froehlich, P.N., jr (1983): Arsenic, antimony, germanium
and tin in the Tejo Estuary, Portugal: Modelling a polluted estuary. Environmental
Science and
Technology, 17, 731-737.
Beek, J. & Van Riemsdijk, W.H. (1979): Chapter 8: Interaction
of orthophosphate ions with soil. In: Bolt, G.H. (ed): Soil
chemistry, volume B: Physico-chemical models. Elsevier,
Amsterdam.
Belzile, N., De Vitre, R.R. & Tessier, A. (1989): Chemistry of
As(V)/As(III) and iron oxyhydroxides in natural lacustrine
sediments. In: Proc. 7th Int. Conf. Heavy Metals Environ.,
Geneva, 512-515.
Belzile, N. & Lebel, J. (1986): Capture of arsenic by pyrite
in near-shore marine sediments. Chemical geology, 54, 279-281.
Belzile, N. & Tessier, A. (1990): Interactions between arsenic
and iron oxyhydroxides in lacustrine sediments. Geochimica et
Cosmochimica Acta, 54, 103-109.
Bowden, J.W. (1973): Models for ion adsorption on mineral
surfaces. Ph.D. Thesis, University of Western Australia.
Brannon, J.M. & Patrick, W.H. (1987): Fixation, transformation
and mobilization of arsenic in sediments. Environmental
Science and Technology, 21, 450-459.
Brooks, R.R., Fergusson, J.E., Holzbecher, J., Ryan, D.E.,
Zhang, H.F., Dale, J.M. & Freedman, B. (1982): Pollution by
arsenic in a gold mining district in Nova Scotia. Environ.
Pollut. Ser. B, 4, 109-117.
Bryan, G.W. & Gibbs, P.E. (1983): Heavy metals in the Fal
estuary, Cornwall: A study of long-term contamination by
mining waste and its effect on estuarine organisms. Occasional
publicaltion No. 2. Mar. Biol. Assoc., U.K.
Bryan, G.W., Langston, W.J., Hummerstone, L.G. & Burt, G.R.
(1985): A guide to the assessment of heavy metal contamination
in estuaries sing biological indicators. Occasional
publication No. 4. Mar. Biol. Assoc., U.K.
Chau, Y.K. & Wong, P.T.S. (1978): Occurence of biological
methylation of elements in the environment. A.C.S. Symp. Ser.,
92, 39-53.
Craig, P.J. & Moreton, P.A. (1985): The role of speciation in
mercury methylation in sediments and water. Environ. Pollut.
B., 10, 141-158.
Crecelius, E.A., Bothner, M.H. & Carpenter, R. (1975):
Geochemistries of arsenic, antimony, mercury and related
elements in sediments of Puget Sound. Environmental Science
and Technology, 9, 325-333.
De Haan, F.A.M. (1965). The interaction of certain inorganic
anions with clays. Agr. Res. Rep. Wageningen, 655.
Ferguson, J.F. & Gavis, J. (1972): A review of the arsenic
cycle in natural waters. Water Research, 6, 1259-1274.
Freeman, M.C., Aggett, J. & O'Brien, G. (1986): Microbial
transformations of arsenic in Lake Ohakuri, New Zealand. Water
Research, 20, 283-294.
Froelich, P.N., Kaul, L.W., Byrd, J.T., Andreae, M.O. & Roe,
K.K. (1985): Arsenic, barium, germanium, tin, dimethylsulfide
and nutrient biogeochemistry in Charlotte Harbor, Florida, a
phosphorus-enriched esturary. Estuarine, Coastal and Shelf
Science, 20, 239-264.
Gillot, J.E. (1968): Clay in engineering geology, Elsevier,
Amsterdam.
Hare, L., Campbell, P.G.C., Tessier, A. & Belzile, N. (1989):
Gut sediments in a burrowing mayfly (Ephemeroptera, Hexagenia
limbata): their contribution to animal trace element burdens,
their removal, and the efficacy of a correction for their
presence. Canadian Journal Fish. Aquat. Sci., 469, 451-456.
Holm, N.G. (1988): Arsenic regeneration from estuarine
sediments of the Bothnian Bay, Sweden. Chemical Geology, 68,
89-98.
Holm, T.R., Anderson, M.A., Iverson, D.G. & Stanforth, R.S.
(1979): Heterogenous interactions of arsenic in aquatic systems. In: Jenne, E.A. (ed):
Chemical modeling in aqueous
systems, American Chemical Society, Washington DC, 162-190.
Huang, W.W., Martin, J.M., Seyler, P., Zhang, J. & Zhong, X.M.
(1988): Distribution and behaviour of arsenic in the Huang He
(Yellow River) Estuary and Bohai Sea. Marine Chemistry, 25,
75-91.
Johnson, D.L. (1972): Bacterial reduction of arsenate in sea
water. Nature, 240, 44-45.
Johnson, D.L. & Pilson, M.E.Q. (1975): The oxidation of arsenite in seawater.
Environmental letters, 8, 157-171.
Jurinak, J.J. & Griffin, R.A. (1972): Nitrate ion adsorption
by calcium carbonate. Soil Science, 113, 130-135.
Kersten, M. (1988): Part two: Geochemistry of priority pollutants in anoxic sludges. In:
Salomons, W. & Förstner, U.:
Chemistry and biology of solid waste. 189-195.
Langston, W.J. (1980): Arsenic in U.K. estuarine sediments and
its availability to benthic organisms. Journal of the Marine
Biology Association, U.K., 60, 869-881.
Langston, W.J. (1983): The behavior of arsenic in selected
United Kingdom estuaries. Canadian Journal Fish. Aquat. Sci.,
40, 143-150.
Lasaga & Kirkpatrick (1983): Chapter 1: Kinetics of
geochemical processes. In: Reviews in mineralogy, volume 8.
Linder, H.R., Seltner, H.D. & Schreiber, B. (1978): Use of
dibenzyldithiocarbaminate as coprecipitant in the routine
determination of 12 heavy metals in pharmaceuticals by X-ray
fluorescence spectroscopy. Analytical Chemistry, 50, 896-899.
Maher, W.A. (1984): Mode of occurence and speciation of arsenic in some pelagic and
estuarine sediments. Chemical
Geology, 47, 333-345.
Massee, R., Maessen, F.J.M.J. & De Goey, J.J.M. (1981): Losses
of silver, arsenic, cadmium, selenium and zinc traces from
distilled water and artificial sea-water by sorption on
various container surfaces. Analytica Chimica Acta, 127, 181-
193.
McBride, B.C., Merilees, H., Cullen, W.R. & Pickett, W.
(1978): Anaerobic and aerobic alkylation of arsenic. In:
Brinckman, F.E. & Belkema, J.M (eds): Organometal and
organometalloids, occurence and fate in the environment.
A.C.S. Symp. Ser., 82, 94-115.
NRCC (1978): Effects of arsenic in the Canadian environments.
Publication No. NRCC 15391, National Research Council, Ottawa.
Oscarson, D.W., Huang, P.M. & Liaw, W.K. (1980): The oxidation
of arsenite by aquatic sediments. J. Environ. Qual., 9, 700-
703.
Oscarson, D.W., Huang, P.M. & Liaw, W.K. (1981a): Role of
manganese in the oxidation of arsenite by freshwater lake
sediments. Clays and Clay Minerals, 29, 219-225.
Oscarson, D.W., Huang, P.M., Defosse, C. & Herbillon, A.
(1981b): Oxidative power of Mn(IV) and Fe(III) oxides with
respect to As(III) in terrestrial and aquatic environments.
Nature, 291, 50-51.
Oscarson, D.W., Huang, P.M. & Hammer, U.T. (1983a): Oxidation
and sorption of arsenite by manganese dioxide as influenced by
surface coatings of iron and aluminium oxides and calcium
carbonate. Water, Air and Soil Pollution, 20, 233-244.
Oscarson, D.W., Huang, P.M. Liaw, W.K. & Hammer, U.T. (1983b):
Kinetics of oxidation of arsenite by various manganes
dioxides. Soil Science Society American Journal, 47, 644-648.
Pannekoek, A.J. (1982): Hoofdstuk 18: Verwering en bodemvorming. In: Pannekoek,
A.J. & Van Straaten, L.M.J.U.: Algemene
Geologie. Wolters-Noordhoff, Groningen.
Penrose, W.R. (1974): Arsenic in the marine and aquatic environments: analysis,
occurence and significance. C.R.C.
Critical Reviews in Environmental Control, 4, 465-482.
Salomons, W. & Förstner, U. (1984): Metals in the hydrocycle.
Springer Verlag, Berlin.
Van der Weijden, C.H. & Middelburg, J.J. (1989):
Hydrogeochemistry of the river Rhine: long term and seasonal
variability, elemental budgets, base levels and pollution.
Water Research, 23, 1247-1266.
Van Elteren, J.T., Das, H.A., De Ligny, C.L. & Agterdenbos, J.
(1989): Determination of arsenic (III/V) in aqueous samples by
neutron activation analysis after sequential coprecipitation
with dibenzyldithiocarbamate. Analytica Chimica Acta, 222,
159-167.
Vidal, F.V. & Vidal, V.M.V. (1986): Arsenic metabolism in
marine bacteria and yeast. Marine Biology, 60, 1-7.



Titel rapport:
